ECFG 9 Parallel Session 5 Secondary Metabolism
PS5.1
Hybrid polyketide synthase - non-ribosomal peptide synthetases (PKS-NRPS) in fungi
Russell Cox, Colin Lazarus, Andrew Bailey, Thomas Simpson, Zhongshu Song, Laura Halo
University of Bristol, Bristol, United Kingdom
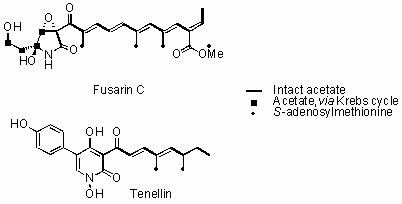
The genomes of filamentous fungi contain numerous gene clusters for the biosynthesis of complex secondary metabolites. These usually include polyketide synthases (PKS), non-ribosomal peptide synthetases (NRPS) and terpenoid synthases, but recently we have shown that hybrid PKS-NRPS systems are relatively common and responsible for the biosynthesis of diverse compounds known as acyl tetramic acids. We have worked with PKS-NRPS genes involved in the biosynthesis of Tenellin (Beauveria bassiana) and Fusarin-C (Fusarium venenatum). These 12Kb ORFs are difficult to manipulate using traditional 'cut and paste' approaches so yeast recombination, combined with GATEWAY in vitro recombination has been used to rapidly assemble expression vectors. Expression in Aspergillus oryzae has been achieved and the chemical analysis of the compounds produced reveals much about the programming of the PKS-NRPS proteins. Co-expression and knockout strategies have revealed more about the post-assembly reactions and have allowed us to probe the wider gene clusters.
PS5.2
Genome mining for fungal secondary metabolites
Axel A. Brakhage1, Julia Schuemann2, Sebastian Bergmann1, Kirstin Scherlach2, Volker Schroeckh1, Christian Hertweck2
1Molecular and Applied Microbiology, Leibniz-Institute for Natural Product Research and Infection Biology (HKI), and Friedrich Schiller University, Jena, Germany, 2Biomolecular Chemistry, Leibniz-Institute for Natural Product Research and Infection Biology (HKI), and Friedrich Schiller University, Jena, Germany
Mixing both genomic data and analytical techniques can be a powerful approach to the discovery of novel and potentially bioactive natural products. Today, if the genome sequence is at hand, it will be possible to estimate the biosynthesis potential for a given organism by mining the whole genome for typical secondary metabolite biosynthesis genes. This approach is particularly promising in microorganisms because most of the natural product biosynthesis genes are organized in clusters. The formation of many important fungal secondary metabolites, polyketides and non-ribosomal peptides involves multifunctional enzymes: polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs). Sequence alignments between genomes of different Aspergilli suggest that Aspergillus nidulans has the potential to generate up to 32 polyketides, 14 nonribosomal peptides, and 2 indole alkaloids. This high number of putative metabolites is greater than the known metabolites ascribed to these species, and it may be a reflection of incomplete natural product analysis in these species or failure of many clusters to be expressed, at least under the culture conditions commonly used in laboratories. We reported a new strategy for the successful induction of a silent metabolic pathway in A. nidulans, which led to the discovery of novel PKS-NRPS hybrid metabolites, named aspyridones A and B. In a broad bioactivity screening they exhibited moderate cytotoxic activities. The function of the aspyridones in the natural context and possible triggers for the onset of their biosynthesis remain the subject of ongoing investigations. However, the results shown here provide the proof of principle for a strategy that may be generally applicable to the activation of silent biosynthesis gene clusters, in particular in eukaryotes, which will lead to the discovery of many so far unknown products and therefore represents a novel avenue to drug discovery. Furthermore, using novel tools we try to identify natural triggers of secondary metabolism gene clusters.
Bergmann, S., J. Schuemann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck (2007) Nature Chemical Biology 3: 213-217
PS5.3
Expression and peroxisomal targeting of the IAT (Isopenicillin N Acyltransferase) and IAT-like enzymes of Penicillium chrysogenum
Carlos Garcia-Estrada1, Inmaculada Vaca1, Ricardo V. Ullan1, Ramiro P. Godio1, Fernando Teijeira2, Juan Francisco Martin1
1Instituto de Biotecnologia (INBIOTEC), Leon, Spain, 2Area de Microbiologia, Departamento de Biologia Molecular, Facultad de CC. Biologicas y Ambientales, Universidad de Leon, Leon, Spain
Introduction: Penicillium chrysogenum and Aspergillus nidulans are filamentous fungi capable of converting isopenicillin N (IPN) into hydrophobic penicillins by means of the penDE-encoded IPN acyltransferase (IAT), a peroxisomal enzyme that requires self-processing to become active. The genomes of P. chrysogenum and A. nidulans also contain a gene (penDE-like) encoding a protein paralogous of IAT, that we have termed IAT-like. Surprisingly, the genome of several penicillin non-producer ascomycetes includes a gene similar to the penDE-like gene. In this work we characterize the penDE-like gene of P. chrysogenum and study the role of the IAT-like enzymes in antibiotic production.
Methods: Site-directed mutagenesis has been used to generate functional peroxisomal targeting sequences (PTS1) in the penDE-like gene. Strong promoters assured gene overexpression. HPLC has been used to determine 6-APA, IPN and benzylpenicillin.
Results: The IAT-like contains important residues included in the wild-type IAT, such as the Gly102-Cys103 processing site. However, the peroxisomal targeting motif was absent in the C-terminus of the protein. The expression of the gene encoding this protein (penDE-like) was tested in the P. chrysogenum Wis54-1255 strain, but no transcription was detected. To test the potential role of IAT-like in penicillin biosynthesis, the penDE-like gene was overexpressed using a strong promoter in the npe10-AB-C strain, which contains the pcbAB and pcbC genes, but lacks the wild-type IAT. No acyltransferase (benzylpenicillin forming) or amidohydrolase (6-APA releasing) activities were detected. To discard that the lack of activity is a consequence of mislocalization, the ARL motif (for peroxisomal targeting) was included in the C-terminus of the IAT-like (IAT-likeARL) by site-directed mutagenesis. The gene encoding IAT-likeARL (penDE-likeARL) was overexpressed in the npe10-AB-C strain, but neither benzylpenicillin nor 6-APA were detected.
Discussion: The lack of transcription of the penDE-like gene, indicates that this gene might be a silent gene. Overexpression and the lack of activity of the IAT-like and IAT-likeARL indicate that even a correct targeting of this protein does not lead to penicillin-related enzymatic activity. The presence of the penDE-like gene in several ascomycetes point out to this gene as an ancestor (of still unknown function) of the wild-type penDE gene.
PS5.4
Secondary metabolism genes in Botrytis cinerea: a high potential for sesquiterpenes biosynthesis
Muriel Viaud1, Isidro Collado2, Jean-Marc Pradier1, Christina Pinedo2, Mathias Choquer & the Botrytis/Sclerotinia Genome Consortium1
1INRA, Versailles, France, 2University of Cadiz, Puerto Real, Spain
Botrytis cinerea is responsible for grey mould on more than 200 plant hosts including grapevine. This species and the closely related Sclerotinia sclerotiorum are the first Leotiomycetes, but also among the first polyphagous and necrotrophic fungi to be sequenced (http://www.genoscope.cns.fr/ & http://www.broad.mit.edu/annotation/fgi/). Comparative genomics may unravel the differences that could be associated with their necrotrophic lifestyles and their broad host ranges.
Secondary metabolism genes were investigated by looking for conserved domains of the relevant « key enzymes »: Terpene cyclases, Polyketide Synthases, Non-Ribosomal Peptide Synthases and Dimethylallyl Tryptophan Synthetases. Candidate genes were investigated by reverse genetics and their distribution was studied in other Botrytis species that, in contrast to B. cinerea, have narrow host ranges.
Genome annotation revealed that B. cinerea and S. sclerotiorum have 41 and 28 key enzymes respectively, and share only half of them. Most genes are organized in physical clusters, and species-specific clusters correspond to synteny breaks suggesting recent cluster gains or losses. The most significant difference is an enrichment of sesquiterpene cyclases (STC) in B. cinerea as compared to S. sclerotiorum (6 versus 1). Functional analysis of the STC genes has been initiated by gene inactivation. STC1/CND15 was shown to be the cyclase necessary for the biosynthesis of botrydial, a toxin that acts as a strain-dependant virulence factor. The STC2 amino acid sequence is similar to the trichothecene cyclase TRI5. Biochemical characterization of STC2/TRI5 null mutants did not allow yet to identify the corresponding metabolite. However, STC2/TRI5 null mutants showed a reduced colonization on bean and tomato leaves.
Southern blot analysis indicated that Botrytis species with narrow host ranges have only few homologs (ranging from 0 to 3) of the 6 STCs identified in B. cinerea.
Altogether genomic and functional data suggest that the polyphagous fungus B. cinerea has acquired a high potential for sesquiterpene biosynthesis as compared to other Botrytis species and that at least two STCs are involved in virulence on different hosts.
PS5.5
Variation in TRI1 in thirteen trichothecene-producing species of Fusarium: evidence for a complex evolutionary history of a mycotoxin biosynthetic locus
Robert Proctor, Susan McCormick, Nancy Alexander, Anne Desjardins
United States Department of Agriculture, Agriculture Research Service, National Center for Agricultural Utilization Research, Peoria, Illinois, United States
Trichothecenes are mycotoxins produced by several genera of fungi, including some agriculturally important Fusarium species. In the two species, Fusarium graminearum and F. sporotrichioides, that have been examined most thoroughly, trichothecene biosynthetic enzymes are encoded at three loci: 1) the 12-gene, core trichothecene biosynthetic gene (TRI) cluster, 2) the TRI1/TRI16 locus, and 3) the TRI101 locus. In F. graminearum, the cytochrome P450 monooxygenase encoded by TRI1 catalyzes trichothecene C-7 and C-8 hydroxylation, whereas in F. sporotrichioides, the enzyme catalyzes only C-8 hydroxylation. Here, nucleotide sequence analysis of 13 Fusarium species distinguished four transpecies groups of TRI1. Within each group, predicted TRI1 amino acid sequences are 85-99% identical, and between groups, sequences are only 65-75% identical. TRI1 sequence variation is not correlated with phylogenetic relationships of Fusarium species inferred by maximum parsimony analysis of nucleotide sequences of beta-tubulin, translation elongation factor, and two core TRI cluster genes. Sequence analysis of TRI1-flanking regions indicated that TRI1 exists in at least three distinct genetic environments. In contrast to variation in TRI1 sequence, these genetic environments are highly correlated with inferred phylogenetic relationships of Fusarium species. Together, these and other data indicate that the evolutionary history of the TRI1/TRI16 locus has been complex and has involved ancestral polymorphism as well as gene relocation, inversion and nonfunctionalization.
PS5.6
Global regulation of bikaverin biosynthesis in Fusarium fujikuroi
Philipp Wiemann1, Marita Beyer2, Hans-Ulrich Humpf2, Bettina Tudzynski1
1Institute of Botany, WWU Münster, Münster, Germany, 2Institute of Food Chemistry, WWU Münster, Münster, Germany
The phytopathogenic ascomycete Fusarium fujikuroi produces different secondary metabolites, including gibberellins (GAs) and the red polyketide pigment bikaverin, which was lately shown to act cytotoxically on several human cancer cell lines. The polyketide synthase gene bik1 was previously shown to be responsible for the first biosynthetic step forming an unmodified nonaketide. Here we present cloning and functional characterization of five new genes, bik2 to bik6; all organized in a bikaverin gene cluster. Furthermore, we studied the complex regulation of these genes in response to different environmental conditions such as nitrogen concentration, pH-value and different growth stages.
The genes bik2, bik3, bik5 and bik6 encode a FAD-dependent monooxygenase, an O-methyltransferase, a Zn(II)2Cys6 transcription factor and a MFS transporter, respectively. Bik4 belongs to the family of NmrA-like proteins but its role in bikaverin production is not clear yet. HPLC-MS analysis of deletion mutants of all bik genes showed loss or reduction of the red pigment, demonstrating that they encode proteins involved in bikaverin biosynthesis. Northern blot analysis revealed that expression of all bik genes is strongly dependent on the cluster specific zinc finger transcription factor Bik5. Beyond this pathway specific regulation, expression of all genes is repressed by high amounts of nitrogen and basic pH. We show that the general transcription factor AreA is not essential, but might be indirectly involved in nitrogen regulation of bikaverin biosynthesis. Deletion of pacC, encoding the Cys2His2 zinc finger regulator PACC, resulted in de-repression of the bik genes under alkaline pH conditions indicating that PACC acts as a repressor. Furthermore, expression analysis of the bikaverin biosynthetic genes in time course experiments showed a downregulation of these genes after 3 days. Deletion of the "velvet" homologue gene veA resulted in an expression of the bik genes up to the 10th day. In contrast, overexpression of veA let to a downregulation of the bik genes even at early time points. Interestingly, the GA genes are regulated by VeA in the opposite way. Our results reveal new aspects of different regulation patterns for genes organized in one cluster in F. fujikuroi, responsible for biosynthesis of the cytotoxic bikaverin, and make this gene cluster an excellent model for regulation studies of secondary metabolism in an ascomycete.
PS5.7
Subcellular localisation of two pathway-specific geranylgeranyl diphosphate synthases in the filamentous fungus Penicillium paxilli
Sanjay Saikia, Barry Scott
Centre for Functional Genomics, IMBS, Massey University, Palmerston North, New Zealand
In the filamentous fungi Penicillium paxilli, Neotyphodium lolii, Fusarium fujikuroi, and Aspergillus flavus two distinct geranylgeranyl diphosphate (GGPP) synthases, one for the primary metabolism (P. paxilli Ggs1-related proteins) and the other for secondary metabolism (P. paxilli PaxG-related proteins) are present. The genomes of other filamentous fungi including A. clavatus, A. fumigatus, A, nidulans, A. niger, A. oryzae, A. terreus, F. graminearum, and Neosartoya fischeri also contain two or more copies of genes homologous to paxG, although the secondary metabolite phenotype of most of these fungi are not known. To understand the biological significance of the presence of multiple copies of GGPP synthases across these genera we are using P. paxilli as our model organism in conjunction with reporter fusion vectors, deletion and complementation analyses. We found that the presence of ggs1 failed to complement a paxG-deletion mutant strain. These observations suggest that the two metabolic pathways are compartmentalised. To test this, reporter fusion studies were conducted and found that Ggs1-EGFP fusion protein was localised to punctuate structures and EGFP-GRV fusion protein, containing the C-terminal tripeptide GRV of PaxG, was localised to peroxisomes. These and more recent findings will be presented.
PS5.8
Genomic insights into chemical diversity of opportunistic human pathogen, Penicillium marneffei, and its close saprophytic relative, Talaromyces stipitatus
William Nierman1, Nora Khaldi2, Fayaz Seifuddin1, Matthew Fisher3, Alex Andrianopoulos4, Geoff Turner5, Natalie Fedorova1
1J. Craig venter Institute, Rockville, MD, United States, 2Smurfit Institute, University of Dublin, Trinity College, Dublin 2, Ireland, 3Imperial College, St Mary's Hospital, Norfolk Place, London W2 1PG, United Kingdom, 4University of Melbourne, Victoria, Australia, 5The University of Sheffield, Sheffield S10 2TN, United Kingdom
Fungi produce a variety of biologically active small molecules or secondary metabolites, which can be toxic (e.g. aflatoxin) or beneficial (e.g. penicillin) to humans. Secondary metabolites are particularly abundant in soil-dwelling filamentous fungi, which exist as multicellular communities competing with each other for nutrients, minerals and water and trying to avoid predation by amoebas, insects or other higher eukaryotes. In fungal genomes, genes responsible for secondary metabolite (SM) biosynthesis, export, and transcriptional regulation are often found in well-defined, contiguous regions or clusters. Here we use this feature to predict SM biosynthetic pathways in recently sequenced genomes of the opportunistic human pathogen, Penicillium marneffei, and its close sexual non-pathogenic relative, Talaromyces stipitatus.
To systematically predict SM pathways in fungal genomes, we have developed a web-based SMURF software tool (available at http://www.jcvi.org/cms/research/software/). We have applied SMURF to catalogue putative SM clusters in the genomes of P. marneffei and T. stipitatus and other publicly available fungal genomes. Initial predictions were based on a gene’s chromosomal position with respect to "backbone" genes (NRPSs, PKSs, hybrids, or DMATs), which encode the first step in SM biosynthesis. Initial clusters were further refined based on PFAM and TIGRFAM domain content.
SMURF analysis has revealed that P. marneffei and T. stipitatus contain 539 and 656 putative SM genes organized in 48 and 61 clusters, respectively. Known mycotoxins produced by T. stipitatus include duclauxin, talaromycins, botryodiploidin, and emodin, although none of them has been characterized at the gene level. Even less is known about SM secreted by P. marneffei. Only 36 of these clusters are orthologous, which means that ~30% of P. marneffei and ~50% of T. stipitatus SM clusters are unique to either species. Overall Penicillium and Aspergillus species have the highest number of SM clusters found in fungal species to date and are likely be a rich source of novel natural products.