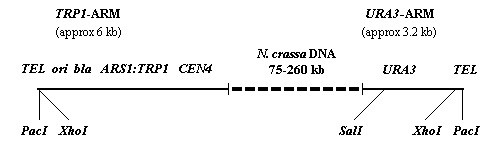
Figure 1. A schematic of a typical clone from the N. crassa YAC library is shown. The markers shown are described in the text.
The following wells in the library contain cultures that will not grow or grow slowly upon inoculation: 1:6A l:llH 2:8F 2:11E 5:9G 6:5F 6:11E 10:1F 10:6A 10:9B l l:lH 11:2G 11:6A 12:7D 12:9C 14:3D 14:3E 14:9E 17:3G 17:6F 17:9H 17:10H 18:3E 19:7G 19:9F 20:2D 20:5E 20:7H 21:4F 22:3D 22:4A 22:9C 23:11D 24:4A 24:4B 24:4C. There are no clones in columns 5 to 12 on dish 24. Additionally, well 21:3E was contaminated with cells from well 21:3D. At least 5% of the clones besides those listed did not grow or grew poorly upon inoculation from a third generation copy of the YAC library distributed by the Fungal Genetics Stock Center (C. Yamashiro, unpublished observations).
Chromosome Walking
1. A protocol is outlined for hybridization screening of the YAC library. Positive
YAC clones obtained from screening the library must be verified by Southern
analysis. Mixed cultures within an individual well can occur; therefore, streak out
cultures for single colonies on selective plates. Prepare genomic DNA from
one or more individual colonies, and perform genomic Southern blot
hybridizations with these DNAs as outlined below.
2. Several of the YAC clones obtained from a given library screening should be physically mapped with restriction endonucleases and the resultant maps compared. They should fit into a single set of contiguously overlapping sequences referred to as a "contig". Construction of a contig map assures the continuity of individual clones and avoids possible confusion resulting from multiple insert clones, whose restriction maps will not fit into the consensus maps generated from the bona fide majority of clones.
3. DNA fragments containing the terminal ends of the inserts of YAC clones which map to the periphery of the contig can be subcloned into E. coli or amplified directly (as described below). These fragments can then be used as hybridization probes for chromosome walking, or RFLP mapping.
Hybridization screening of the YAC library. (B. Brownstein et al. 1989. Science 244: 1348-1351, and B. Brownstein personal communication with modifications)
I. Preparation of a YAC library filter set.
1. Positively charged nylon membranes (MSI Inc, Westboro, MA) are layered
onto the surface of large petri plates containing SD medium with casamino acids
and adenine, "SD + CAA + A" (a supplemented minimal medium). Cultures
from microtiter wells are replicated onto marked membranes using a multi-pin
replicator. Each microtiter plate should be replicated onto a single membrane.
Growth will take 3 to 4 days at 30oC. Fast growing colonies can be allowed to
grow quite large (0.5 mm or larger) to allow the slow growing colonies to be well
established (other protocols prefer smaller colonies so that cells are actively
growing but lysis of old colonies works well here).
2. Prepare spheroplasting buffer CES/DTT/YLE (CDY). For 40 ml: Add 80 mg yeast lytic enzyme, 70,000 units/gram (ICN Biochemicals Irvine, CA) and 300 ml of 2 M dithiothreitol (DTT) to 40 ml CES solution (1 M sorbitol 100 mM Sodium citrate pH 7.0, 50 mM EDTA pH 8.0).
3. Place Whatman 3 MM or equivalent in lid of 150 mm Petri dish. Add approximately 7.0 ml CDY to Whatman and remove any bubbles (only a small amount of excess liquid should be present in lid). Carefully place a membrane containing YAC colonies onto the CDY saturated paper. Carefully remove bubbles from under the membrane.
4. Place the bottom of the Petri dish over the membrane and seal dish well with parafilm. Incubate 2 days 30-32oC.
5. Colony lysis and DNA denaturation is done by placing the membranes sequentially on Whatman 3MM paper saturated with the following solutions at room temperature for the times shown:
a. 10% SDS/100 ug/ml proteinase K for 10 min.
b. 0.5 N NaOH for 10 min.
c. Three washes of 200 mM Tris pH 7.5/2X SSC for 5 min each.
6. Air dry the membranes on 3 MM paper for 2 h minimum (can go overnight). Bake for 2 h in vacuum oven at 80oC. Wrap membranes in plastic wrap (do not stack membranes, arrange them side to side). Store at -20oC.
II. Hybridization.
1. Prehybridize filters overnight at 65oC in a prehybridization (prehyb) solution
of 1 M NaCl, 1% SDS and 10% dextran sulfate (Oncor Inc. Gaithersburg,
MD). Four sets of six filters were each prehybridized with 50 ml of prehyb
solution. Filters were incubated in plastic "Tupperware" containers with the lids
on (hybridization bags could also be used). Discard the prehyb solution if cell
debris is present (lots of cell debris will be present after the first prehybridization
of the filter set).
2. Hybridization: Use 1 to 5 x 10^5 cpm of 32P-labeled probe/ml of prehyb solution. Boil carrier salmon or herring sperm ssDNA (100 ug/ml in final hybridization) with the probe for 10 min to reduce background caused by non- specific hybridization. Add labeled probe with carrier DNA to hyb cocktail in fresh Tupperware containers then add filters from prehybridization. Four sets of six filters were each hybridized with 50 ml of cocktail. Hybridize at 65oC (use from 42oC to 65oC depending on stringency) overnight with shaking. To save money, we hybridized two sets of six filters one night, then reused the hybridization cocktail for the remaining filters the following day.
Note: YAC vector pYAC4 contains the entire pBR322 DNA sequence. Do not use probes with this sequence, otherwise your probe will hybridize to all the YAC clones. Alternatively, a 50-fold molar excess of cold competitor pBR322 DNA can be added to the hybridization reaction to reduce background. Similarly, if labeled cosmid DNA is used as a probe use a 50-fold molar excess of cold cosmid vector (i.e., lacking insert DNA) to decrease background (vector-vector) hybridization.
3. Remove membranes for washes on a gyratory shaker. Wash times should be
adjusted to yield background signal acceptable to you.
a. Wash in 2 changes of 2X SSC/ 1% SDS for 30 min each at 65oC.
b. Wash in 2 changes of 0.5X SSC/ 1% SDS for 30 min at 65oC. For
higher stringency use 0.1X SSC/l % SDS for 1 h at 75oC.
4. Wrap membranes in plastic wrap and expose to film.
5. Rehybridization: We did not strip our library filters. Instead, we prepared two filter sets and each set was allowed to decay approximately 3 months prior to rehybridization. If filters are to be stripped do not allow them to dry out at any time (store wet at -20oC).
Selective medium (SD + CAA +A) 0.67% Bacto-yeast nitrogen base without amino acids, 2% dextrose, 0.5% casamino acids, 15 mg adenine/liter. For solid medium add 2% agar.
YAC restriction mapping and contig building.
Restriction maps of YAC clones can be generated quickly by partial digestion and
indirect end labeling (M. Centola and J. Carbon. 1994. Mol. Cell. Biol. 14:1510-1519) as
follows: Chromosomal DNAs of the YAC clones imbedded in agarose "DNA plugs" are
prepared. DNA plugs are partially digested with a restriction endonuclease which
recognizes an 8 bp DNA sequence. Partial digestion reactions are fractionated on
CHEF
gels and blotted to membranes. Membranes are then hybridized with probes specific
for
one of the YAC vector arms. A series of positive hybridization bands of increasing size
will
be imaged upon autoradiography of the membrane in each lane containing a partial
digestion reaction. The size of each band is calculated relative to size standards run on
the
fractionation gel. The difference in size between sequential bands is equal to the
distance
between restriction sites in a given clone. Resultant restriction maps can be verified by
rehybridizing stripped blots with a probe specific for the opposite arm of the YAC clone.
Restriction maps of individual clones can then be overlapped and contigs can be
generated.
I. Preparation of chromosomal DNA plugs of YAC clones (Kuspa et al.. 1989. Proc. Natl. Acad. Sci. USA 86:8917-8921).
A. Small scale preparation:
1. Grow 3 ml culture of yeast strain in SD+CAA+A medium at 30oC to
stationary phase (2-3 days).
2. Pellet 1.5 ml of cell culture in a microfuge tube and wash pellet with 1 ml 0.5 M EDTA pH 8.0.
3. Remove EDTA solution, respin cells and remove ALL remaining EDTA solution with a pipetman.
4. Gently resuspend cells in 10.8 ul spheroplasting solution.
5. Incubate at room temperature (RT) for 5 min.
6. Add 25.2 ul of molten 1.2% LMP agarose in SCE cooled to 45oC (final concentration of agarose is 0.6%).
7. Pour into plug molds. Solidify at RT for approximately 20 min.
8. Move plugs into microfuge tube. Add overlay solution to fill tube. Incubate overnight at 37oC to produce spheroplasts.
9. Remove overlay solution carefully.
10. Add 1 ml of NDS; incubate for 2 days at 50oC.
B. Large Scale Preparation
1. Grow 50 ml culture of yeast strain in a 125 ml flask, with shaking, to stationary
phase (2-3 days) at 30oC in SD+CAA+A medium; cultures will turn pink.
2. Pellet cells 5 min at approximately 1000 x g.
3. Wash with 100 ml 50 mM EDTA pH 8.0 (EDTA solution).
4. Remove EDTA solution, respin cells and with a pipetman remove ALL remaining EDTA solution.
5. Gently resuspend pellet in 400 ul spheroplast solution.
6. Incubate at RT 5 min: briefly warm cells to 45oC (10-20 sec).
7. Add 800 ul 1.2% LMP agarose in SCE. Cool molten agarose to 45-48oC prior to addition. Mix gently and completely. Quickly pour into plug molds. Solidify at RT for approximately 20 min.
8. Move plugs into 15 ml Corex tube add 12 ml overlay solution. Incubate overnight at 37oC to produce spheroplasts.
9. Remove overlay solution carefully.
10. Add 10 ml of NDS and incubate overnight (for 1-2 days) at 50oC. Plugs can be stored in NDS at 4oC.
Solutions with volumes for large scale preps are given below. Final volumes can be scaled up or down.
Spheroplast solution
1 ml SCE
25 ul 2-mercaptoethanol
1 mg Zymolyase (100T) (ICN Biochemical, CA)
1.2% LMP agarose
0.12 g LMP agarose
10 ml SCE (melt agarose)
Overlay solution
46 ml 0.5 M EDTA
0.25 ml 2M Tris pH 8.0
3.75 ml 2-mercaptoethanol
NDS
1 mg/ml Proteinase K
1% N-lauroyl sarcosine in 0.5 M EDTA pH 9.0
SCE
1 M sorbitol
0.1 M sodium citrate, pH 5.8
10 mM EDTA (filter sterilize)
Plug molds: Use 1 cc syringe barrels with the ends sealed with parafilm or slide holders that come with common microscope slides. Tape up sides of slide holder and pour in molten agarose to form plugs that fit nicely into gel wells.
II. Partial restriction enzyme digestion of YAC DNA plugs.
Plugs must be extensively washed prior to digestion. Wash a given volume of plugs
5 times with 25 volumes of 10 mM Tris pH 8.0, 50 mM EDTA at RT for 2 hr/wash.
Plugs can be washed and then stored in wash solution at 4oC for months prior to
digestion.
1. Add the equivalent of approximately 20 ul volume of solid chromosomal DNA plug/tube to two microfuge tubes (approximately 0.2-1 ug of DNA). Two digestion reactions will be done on each clone.
2. To each tube add 100 ul 0.5 mM phenylmethylsulfonyl fluoride (PMSF; USB, Cleveland, OH) in TE buffer. Incubate reactions at 37oC for 30 min. PMSF is highly unstable in aqueous solution (half life = 30 min); prepare a fresh 100 mM stock in 100% ethanol and dilute to 0.5 mM immediately before each use.
3. Remove PMSF wash solution and add 1 ml of TE buffer to plug. Incubate at 37oC for 30 min and repeat wash once. Remove final wash solution completely.
4. Add 1/10 volume of restriction enzyme buffer (10 x concentrate) to plug and melt mixture at 68oC for 5 min. Plugs should be completely molten.
5. Cool plugs to 37oC and add 0.2 and 1.0 units of pre-warmed (37oC) restriction enzyme to each of the two reactions, respectively. The plug should still be molten. Mix gently with a pipet tip. Incubate at 37oC for 30 min.
6. Add 1 ul of 0.5 M EDTA (pH 8.0) and remelt plugs at 68oC for 5 min and the molten reactions loaded onto an agarose gel (see below).
Note: Complete restriction enzyme digests of the DNA within agarose plugs can be done as described above if a large amount of enzyme is used (we typically use 12-30 units of enzyme/20 ul of plug) and incubate reactions at 37oC for 3 hr or more.
III. Using CHEF gel analysis to resolve YAC clones.
Partial digests are fractionated on a 1% agarose gel in 0.5 x TBE (1 x TBE = 90
mM
Tris-borate, 2 mM EDTA). Gels (10 x 10 cm) are subjected to electrophoresis at 6.0
V/cm
for 15 h in a CHEF gel apparatus (BioRad, Richmond CA), using a 0.2 to 13 sec pulse
ramp, with a 120o pulse angle, at 14oC. Lambda concatemers and a 5 kb ladder
(pBR328
concatemer, BioRad) size standards are also loaded on the gel. Stain gel with ethidium
bromide, and photograph with a ruler atop the gel so migration distances of standards
can
be determined. These conditions will resolve DNA fragments ranging from 5-250 kb and
are thus suitable for both partial digest mapping and for analysis of complete digestion
products by Southern hybridization.
Some YAC clones are >250 kb. Resolution of uncut YAC clones can be done using the following conditions: 1% agarose gels prepared and subjected to electrophoresis in 0.5 x TBE buffer at 6.0 V/cm, using a 6-12 sec ramped switch time, at 12oC for 16-20 hr. These conditions will resolve clones up to 400 kb in size. Plugs can be placed into the wells of a CHEF gel by balancing the plug on a glass coverslip and pushing the plug into the well with a pipet tip. Alternatively, as suggested by Virginia Pollard, plugs can be placed onto the teeth of the well comb and the agarose gel cast with the comb in place. One needs to check for any dislodged plugs which can be repositioned using a Pasteur pipet.
IV. Southern Hybridization.
Southern blotting of high molecular weight DNA requires extensive nicking of the
DNA prior to transfer, and a large volume of transfer buffer to be used during capillary
transfer.
1. Nick high molecular weight DNA within the ethidium bromide stained gel using a commercial UV cross linker. Use settings recommended by the vendor; BioRad crosslinkers have a program for nicking pulse field gels that works nicely. Alternatively, place the gel into 0.2 N HCl for 30 min at RT with gentle agitation.
2. Denature gel in 0.5 M NaOH, 1.5 M NaCl for 30 min.
3. Neutralize in 1 M Tris pH 7.5, 1.5 M NaCl for 30 min.
4. Transfer to a positively charged nylon membrane using standard capillary blotting procedures with the following modifications:
a. Use a very large wick of 3MM paper, approximately 35 cm long x 30
cm wide, under the gel.
b. Use 2 liters of 10x SSC for each transfer. Use a large volume of
absorbent material above the gel. Use two glass plates (approximately
30 cm x 20 cm) as weights above the absorbent, excessive weight will
compress the gel and prematurely stop the transfer.
5. Standard Southern hybridization conditions can be used.
6. Hybridization probes specific for the left and right arms of the pYAC4 vector can be obtained from the following sources:
a. Gel-purified 1.7 kb BamHI-PvuII fragment from pBR322 hybridizes
specifically to the URA3 -encoding YAC vector arm (URA3-arm).
b. Intact pBluescript (Stratagene, La Jolla, CA) hybridizes specifically to
the TRP1- encoding YAC vector arm (TRP1-arm).
Isolation of terminal restriction fragments from cloned DNA inserts in YAC clones.
Terminal restriction fragments of the insert DNA ("terminal fragments")
from the
TRP1-arm can be directly subcloned into E. coli by plasmid rescue and terminal
fragments
from the URA3-arm can be subcloned into E. coli. Alternatively, terminal restriction
fragments from either end of YAC insert DNA can be cloned by ligation-mediated PCR.
Although large regions of DNA (up to 30 kb) can be subcloned by plasmid rescue, the
procedures are involved and at times frustrating. We therefore recommend the use of
ligation-mediated PCR for obtaining small probes for chromosomal walking.
To isolate terminal end fragments of YAC inserts by plasmid rescue: Digest total genomic DNA from a given YAC clone with XhoI, or PacI (a double digest with XhoI and SalI should also work). These enzymes cut the vector arm once on the telomere proximal side of the E. coli bla and ori. These enzymes should also cut somewhere in the insert DNA. The digestion reaction is then diluted and ligated to circularize the DNA fragment containing bla, ori and the terminal restriction fragment of the insert DNA. A portion of the ligation reaction is electroporated into E. coli and AmpR colonies selected and subsequently screened for the rescued plasmid. Very large (>25 kb) plasmids do not rescue well into E. coli; therefore, determine the expected size of the rescue plasmid for a given enzyme before doing a rescue by preparing a Southern blot of CHEF gel fractionated genomic DNA cut with PacI, XhoI or XhoI/SalI from a given YAC clone and probing with a TRP1-arm specific probe. This will yield the approximate size of the terminal restriction fragment to be rescued. This information should be used to ensure that the rescued plasmid is of the correct size.
Similarly for subcloning terminal fragments from the URA3-arm, digest total genomic DNA from a given YAC clone with PacI, XhoI or XhoI/SalI and shotgun clone the entire genome into an E. coli vector (we used pBluescript). The yeast URA3 gene heterologously rescues the Ura phenotype of E. coli strain DB6656 lacZ 624(Am) trp-49(Am) pyrF79::Mu rpsl179 hsdR27 (Bach, et al. 1979 Proc. Natl. Acad. Sci. USA 76:386). The ligation reaction is introduced into this strain by electroporation and Ura+ transformants selected.
I. Preparation of miniprep DNA from liquid cultures of YAC clones (L.
Clarke and M.
Baum, personal communication).
1. Inoculate 10 ml of SD + CAA + A with yeast cells late in the day in a 125
ml or larger flask. Incubate overnight with shaking at 32oC until culture
reaches an OD660 of 0.6-0.7 (higher concentration cultures yield DNA
resistant to restriction enzyme digestion). Cell cultures can be transferred to
50 ml disposable corning tubes upon reaching the proper OD, pelleted
and stored at -20oC for several days before beginning DNA preps.
2. Wash with 2 ml 1 M sorbitol .
3. Loosen cell pellets before addition of SCE by brief vortexing, then resuspend cells gently in 2 ml SCE containing 0.2 mg Zymolyase 100T and 5 ul 2- mercaptoethanol. Incubate at 37oC for 1 h (never vortex the cells once Zymolyase is present).
4. Centrifuge cells at 800 x g for 4 min, and wash pellet with 2 ml SCE.
5. Remove wash, and resuspend cells in 1 ml NE solution (0.15 M NaCl, 0.1 M EDTA pH 8.0) containing 1% SDS. Add 5 ul 10 mg/ml DNAse free RNase, incubate 15 min at 37oC.
6. Add 5 ul proteinase K (20 mg/ml) and incubate another 15 min at 37o
C. 7. Extract with 1.5 ml phenol/CHCl3/isoamyl alcohol (25:24:1). Invert gently 10 times. Lay tubes on their sides for 5 min. Spin 10 min at 1600 g in clinical centrifuge. Remove aqueous layer with P1000 pipetman using pipet tips with cut off ends (cut off ends so orifice is approximately 0.3 cm in width). Interface is prone to mixing with aqueous phase so use caution. Repeat extraction once.
8. To aqueous layer add 0.12 ml of 7.5 M NH4OAc and 1.2 ml isopropanol, invert to mix. Spin at 8500 rpm for 10 min.
9. Wash pellet with 5 ml of 70% ethanol. Air dry pellet, and resuspend in 50 ul TE buffer.
20 ml SCE: 10 ml 2 M sorbitol , 2 ml 1 M Sodium citrate pH 5.8, 2.4 ml 0.5 M EDTA pH 8.0, 5.6 ml H20.
Note: Be aware of the shear forces generated during the resuspension steps. Be gentle but thorough. Use cut off pipet tips when aliquoting DNA.
II. Plasmid rescue of the terminal restriction fragment from the TRP1-arm
(L. Clarke and
M. Baum, personal communication with modifications).
1. Digest 5 ul of miniprep DNA in a total reaction volume of 50 ul with 20-30
units of restriction enzyme for 3 h at 37oC. Miniprep yields will vary -
5 ul will yield about 2-5 ug. (save an aliquot of the digest to run on a
pulse-field gel to confirm complete digestion and expected size as
described above).
2. Add 40 ul H2O + 10 ul 3 M NaOAc + 40 ul phenol/CHCl3/isoamyl alcohol (25:24:1), "PCI" and mix.
3. Microfuge for 5 min to separate phases and transfer aqueous phase to a new tube.
4. Precipitate with 2 volumes of ethanol at -20oC for 1.5 h.
5. Spin in microfuge at full speed for 20 min.
6. Wash pellet with 0.5 ml 70% ethanol.
7. Resuspend pellet in 650 ul TE buffer and reprecipitate with 100 ul 7.5 M NH4OAc + one volume of isopropanol (second precipitation helps to further clean up the reaction).
8. Pellet and wash as above.
9. Resuspend pellet in 1 ml of 1 x T4 DNA ligase buffer and add 2 units of T4 DNA ligase, incubate at room temperature overnight.
10. Split the reaction into two 500 ul aliquots; to each add 50 ul 3M NaOAc + two volumes of ethanol.
11. Store one aliquot at -20oC (back-up reaction) and the other precipitate at -70oC for 20 min.
12. Pellet and wash as before.
13. Resuspend pellet in 25 ul TE buffer.
14. Introduce a 1 ul aliquot of the reaction mix into E. coli by electroporation and select for AmpR colonies. Note: Plasmid rescues containing repeats will tend to delete if ampicillin concentration in the selective media is >35 mg/l. Standard transformations can be used; however, it is more difficult to get rescues with lower transformation efficiencies.
III. Subcloning of terminal restriction fragment from the URA3
arm.
1. Digest 1 ug of miniprep yeast DNA with 20-30 units of restriction enzyme in
a 50 ul final volume for 3 hr at 37oC as described above.
2. Digest 1 ug of pBluescript (or appropriate vector) with 20 units of enzyme as in step 1.
3. Add 0.5 unit of calf intestinal alkaline phosphatase (Boehringer Mannheim, Indianapolis, IN) to the pBluescript reaction, incubate an additional 20 min at 37oC.
4. Extract each reaction with an equal volume of PCI.
5. Precipitate DNAs with 1/10 volume of 7.5 M NH4OAc + 1 volume of isopropanol.
6. Resuspend each reaction in 10 ul TE buffer, and check the DNA concentration by gel electrophoresis. (Alternatively, if you feel lucky assume your yield and proceed).
7. Ligate 100 ng of digested genomic DNA and 100 ng of prepared vector in a 20 ul reaction volume with 1-2 units of T4 DNA ligase overnight at 16oC.
8. Introduce 1 ul of the ligation reaction into E. coli strain DB6656 by electroporation and select for Ura+ transformants on E. coli minimal medium supplemented with 50 ug/ml tryptophan, and 50 ug/ml ampicillin (T. Maniatis et al. 1982. Molecular cloning, N.Y.). Colonies will appear after 2-3 days.
IV. Ligation-mediated PCR.
Ligation-mediated PCR (LM-PCR; Mueller and Wold 1989. Science 246: 780-786)
is a procedure to isolate DNA fragments for which the sequence of the DNA is
unknown.
This technique has been adapted for generating DNA fragments corresponding to the
end
of inserts within YAC clones (Kere et al. 1992 Genomics 14: 241-248). Below we
describe
the LM-PCR technique used for generating end fragments of inserts within YAC clones
with
several minor modifications.
1. Six oligonucleotides are needed (5' to 3'):
L primer further from the cloning site (oligo L1)
CACCCGTTCTCGGAGCACTGTCCGACCGC
R Primer further from the cloning site (oligo R1)
ATATAGGCGCCAGCAACCGCACCTGTGGCG
L primer nearer the cloning site (oligo L2)
TCTCGGTAGCCAAGTTGGTTTAAGG
R primer nearer the cloning site (oligo R2)
GTCGAACGCCCGATCTCAAGATTAC
Linker long strand and linker primer
GCGGTGACCCGGGAGATCTGAATTC
Linker short strand
GAATTCAGATC
2. Digest 1 ug of yeast DNA embedded in agarose plugs (approximately 20 ul
of plug) to completion as described above using restriction enzymes leaving
blunt ends, such as RsaI, AluI, PvuII, EcoRV, or ScaI (usually set up 2-4
different digests for each clone). One can also digest 1 ug of total
genomic DNA obtained from the miniprep procedure (above) in a 20 ul
reaction volume.
3. Use 5 ul of the restriction digest and ligate the linker by adding ligation buffer, 25 pmol linker, and 1-2 units of T4 DNA ligase in a total reaction volume of 20 ul. Leave at room temperature for >1 h.
4. Set up the PCR using 2-5 ul ligation reaction, 20 pmol long vector-specific primer (L1 or R1) and 10 pmol of linker primer in a total volume of 50 ul. Cycling conditions are 94oC for 1 min, 65oC for 2 min, and 72oC for 2 min for 30-35 cycles.
5. Visualize products by subjecting 5-10 ul to electrophoresis on a 1.5% agarose gel.
6. To make probes from these fragments (they still have significant amount of vector sequence at this time), use PCR amplification of the purified first PCR product with 20 pmol of short vector-specific primer (L2 or R2) and 10 pmol of linker primer in a total volume of 50 ul with the same cycle conditions used in step 4. Purifying the first PCR product is simple by first coring out a small amount of agarose containing the band of interest with a pasteur pipet and transferring to a microfuge tube. Add 100 ul of TE buffer and boil for 5 min. Use 1 ul for the second LM-PCR reaction. The PCR reaction can be done in the presence of 32P-nucleotide for a radioactively labeled probe or in the presence of digoxigenin labeled dUTP (Boehringer Mannheim) for a non-radioactive probe (the latter probe can be used according to protocols provided by Boehringer Mannheim with the Genius kits and systems for non-radioactive detection of nucleic acids).
Transformation of N. crassa with a YAC clone.
I. Introduction of a N. crassa selectable marker into a YAC clone.
The pYAC4 vector does not encode a N. crassa selectable marker. Therefore, transformation of a given YAC clone is limited to clones that encompass selectable markers and must be carried out in appropriate mutant strains. Introduction of a clone from the library into N. crassa has been completed by transforming a qa-2, arom-9, inl strain of N. crassa (FGSC #3952) with total genomic DNA from YAC clone AB1380/YAC 12-10-H (M. Centola unpublished observations). This clone encompasses the qa-2+ gene. Standard spheroplast transformation procedures were used (Vollmer and Yanofsky 1986. Proc. Natl. Acad. Sci. USA 83:4869-4873) and 10 transformations using 1 ug of DNA/150 ul of competent cells yielded a single transformant.
To facilitate introduction of any YAC clone in the library into N. crassa a yeast integration vector carrying a dominant N. crassa selectable marker was constructed. The plasmid pLUShph (diagrammed in figure 2A) encodes the hygromycin phosphotransferase gene (hph) under the control of a modified cpc-1 regulatory region (Royer and Yamashiro 1992. Fungal Genetics Newsl. 39:76-79). This plasmid (pLUShph) also contains the yeast LYS2 gene, and an E. coli KanR gene and ori. This plasmid was constructed by inserting the 3.0 kb HindIII fragment from pMP6 into the unique HindIII site on pLUS (Hermanson et al 1991. Nucl. Acids Res. 18:4943-4948).
Integration of the pLUShph vector into the YAC clones
This plasmid can be site-direct integrated into the URA3-arm of the YAC clones by
digesting pLUShph at the unique SalI site and introducing the linearized plasmid into
the
yeast host strain (diagrammed in figure 2B).
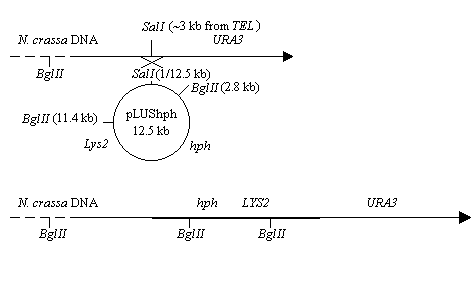
Figure 2. YAC integration vector pLUShph. A) A schematic of the YAC integration vector is shown. The region designated sup4-URA3 is homologous to a portion of the URA3-arm of the YAC clones. As described in the text, this region can be utilized to mediate site-specific integration of this vector into the URA3-arm of any YAC clone. B) A schematic of the integration event is shown. The product of integration contains the yeast LYS2 gene for phenotypic selection of integrated clones. A typical YAC integrant, shown at the bottom of the figure, also carries the N. crassa dominant selectable marker hph that can be used for selection of the YAC integrants upon introduction into N. crassa.
We have integrated this plasmid into several YAC clones by transforming competent yeast spheroplasts. However, lithium acetate transformations (Elble 1992. BioTechniques 13:18-20) are much easier to perform and transformation of LiOAc competent yeast cells with 1 ug of pLUShph linearized with SalI should also be successful. The integrants are selected on dropout media lacking lysine and tryptophan (see below). The DNA from the colonies displaying the Lys+ phenotype was prepared in agarose plugs.
Dropout medium - SD + Adenine + essential amino acids (no lysine)
0.67% Bacto-yeast nitrogen base without amino acids
2% Dextrose
15 mg adenine/liter
2% agar.
Supplemented with the following amino acids. Final concentrations are shown in
ug/ml.
aa (final conc.)
Arg (20)
Asp (100)
Met (20)
Glu (100)
Phe (50)
Val (150)
His (20)
Ser (375)
Tyr (30)
Leu (60)
Thr (200)
II.Verification of Integration by Southern hybridization and Restriction Enzyme
Analysis.
A. Detection of an integrated copy of the hph gene within YAC clones by
Southern analysis
1. Prepare DNA from the Lys+ yeast colonies in agarose plugs (see above for
plug preparation protocol).
2. Fractionate the undigested DNA from the Lys+ transformants and parental host strains on a CHEF gel (use conditions described above for 5-250 kb resolution).
3. Transfer the DNA to a nitrocellulose membrane.
4. Hybridize the membrane with a radioactively-labeled DNA fragment containing the hph gene. We used the 1.2 kb BamHI/ClaI DNA restriction fragment from pCSN44 that encompasses the hph coding region (available from the FGSC). YAC clones in which pLUShph has integrated will hybridize to the probe.
B. Restriction Mapping of hph containing YAC clones.
1. Digest the DNAs from Lys+ YAC clones that hybridize to the DNA fragment
containing the hph gene and the parental host strains with a restriction
endonuclease which cuts inside pLUShph, but not inside pYAC4 (see
"Restriction enzyme digestion of YAC DNA plugs" protocol shown above).
We used the restriction endonuclease BglII.
2. Fractionate the digested DNAs on a CHEF gel (see "Using CHEF gel analysis to resolve YAC clones" protocol).
3. Transfer the DNA to a nitrocellulose membrane.
4. Probe the Southern blot with a radioactively-labeled DNA fragment which has homology with the YAC arm and pLUShph. We probed the Southern blot with the 1.7 kb BamHI/PvuII fragment from pBR322.
5. Expose the blot to film.
6. From the autoradiograph, determine the distance from the end of the telomere to the first restriction site within the insert of the parental YAC clone.
7. From the known restriction map of pLUShph, determine the size of the restriction fragments one would get if the plasmid were integrated at the unique SalI site. This is done by adding the distance from the end of the telomere to the SalI site on the YAC arm into which the plasmid was integrated to the distance between the SalI site on pLUShph and the first restriction site on the SUP4 side of the plasmid. Conversely, one will see a band with a size corresponding to the distance from the restriction site within the insert to the SalI site on pLUShph to the next restriction site on the URA3 side of pLUShph.
Acknowledgements. We are grateful for the support and encouragement of John Carbon, Charles Yanofsky and David Perkins. We thank David Catcheside, Jack Kinsey, and Ed Cambareri for critically reading the manuscript.