NEUROSPORA 2008 PLENARY SESSION ABSTRACTS
Session I: Gene Silencing, Sex, Repair and Recombination
Beadle and Tatum Award Lecture
Genetic Dissection of Meiotic Silencing by Unpaired DNA
Patrick Shiu. University of Missouri- Columbia
A gene present in an abnormal number of copies is usually a red flag for mischief. One way to deal with these potential intruders is by destroying their transcripts, a phenomenon known as RNA interference (RNAi). Meiotic Silencing by Unpaired DNA (MSUD) is an RNAi-based mechanism that surveys the genome of Neurospora crassa. In this system, genes that are not paired during meiotic prophase I, as well as all homologous copies, are silenced during the sexual phase. MSUD is likely to involve the production of double-stranded RNA, which is mediated by the sad-1-encoded RNA-directed RNA polymerase. Additional genetic loci that are important for the silencing pathway, the interactions among different components of the MSUD machinery, as well as the tools developed for their studies (e.g. Bimolecular Fluorescence Complementation, targeted gene placement), will be discussed.
Sexual development in Neurospora crassa: Elusive recessive mutations
Mary Anne Nelson, Christine Chee, Joseph Kunkel, Diego Martinez, and Charles Sanchez. Department of Biology, University of New Mexico
Knockouts (KOs) of each of the ~10,000 genes of the filamentous fungus Neurospora crassa are being constructed by the NIH-supported Program Project, Functional Analysis of a Model Filamentous Fungus, Jay Dunlap, PI. One of the goals of this project is to characterize phenotypes for each of the genes in N. crassa. The NIH P01 award supports limited vegetative and sexual phenotypic analyses. In a pilot project, we have extended those analyses to include self or homozygous crosses of the KO strains, in order to identify strains with recessive sexual defects. We are screening the KO strains for dominant and recessive sexual development defects, as well as male-specific defects. In preliminary work, about 5% of the KO strains made no protoperithecia (female sexual structures), and 3% made no fruiting bodies (perithecia). One KO strain formed perithecia but no sexual progeny (ascospores). The challenges for extending this analysis to include all Neurospora genes will be discussed.
Getting connected: Formation and function of the hyphal network
Louise Glass. University of California, Berkeley
Hyphal fusion occurs at different stages in the vegetative and sexual life cycle of filamentous fungi, such as Neurospora crassa. Similar to cell fusion in other organisms, the process of hyphal fusion requires cell recognition, adhesion and membrane merger. Cell fusion during vegetative growth in N. crassa is a self-signalling process that occurs between both genetically identical and genetically dissimilar germlings/colonies. Analysis of fusion mutants in N. crassa has revealed that genes previously identified as having non-fusion related functions in other systems, have novel hyphal fusion functions in N. crassa. We have focused on a MAP kinase, MAK-2, and SO, a protein of unknown molecular function, which are required for hyphal/germling fusion in N. crassa. Subcellular localization of GFP fusion constructs showed that both proteins localize to the tips of germling fusion cells during chemotropic interactions. We constructed a mak-2 allele whose kinase activity can be specifically inhibited by addition of inhibitor. Disruption of MAK-2 kinase activity by drug addition disturbs the signalling process between MAK-2 and SO. Localization and inhibitor studies also suggest that kinase activity of MAK-2 is required for late fusion events, while SO is not. Understanding the molecular basis of cell fusion in filamentous fungi provides a paradigm for cell communication and fusion in eukaryotic organisms. Furthermore, the physiological and/or developmental role of hyphal fusion is not understood in these organisms; identifying these mechanisms will provide insight into environmental adaptation.
Recent advances understanding the control of DNA methylation
Eric U. Selker. Institute of Molecular Biology, University of Oregon, Eugene, OR 97403. selker@uoregon.edu
We are interested in better defining the mechanisms that control the distribution of DNA methylation in eukaryotic genomes. Twenty-five years ago, while in Bob Metzenberg’s laboratory, I discovered DNA methylation in Neurospora crassa and decided to exploit this organism to explore its function and control. Other model lower eukaryotes, including yeasts, Drosophila and C. elegans showed no methylation. Our early work led to the discovery of RIP (the first described genome surveillance mechanism in eukaryotes), identified sequences that trigger DNA methylation and demonstrated that DNA methylation can interfere with transcriptional elongation. Analyses of the genomic distribution of DNA methylation revealed that it is almost exclusively in relics of RIP, which are mostly in the centromeric regions but are also found near telomeres and at scores of dispersed sites. In addition the tandemly repeated rDNA is lightly methylated. Throughout our studies we have used traditional genetic methods to identify components of the DNA methylation machine and this approach has become increasingly fruitful, especially in conjunction with biochemical and proteomic methods. The availability of the genome sequence and the development of methods to carry out reverse genetics has also accelerated our progress. Previously we reported the identification of several key players in DNA methylation, including a DNA methyltransferase (DIM-2), a histone H3 lysine-9 methyltransferase (DIM-5), a homolog of heterochromatin protein-1 (HP-1) and histone deacetylases (HDACs). Recently we developed an efficient method to identify additional methylation mutants and have placed 14 new mutants into five new complementation groups. In addition, we have applied a new microarray-based method to detect restriction site polymorphisms and used this to map the mutations in some of the new dim (defective in DNA methylation) mutants. I will describe several newly identified dim genes that we have found to be essential for DNA methylation and will present a model summarizing our current understanding. I will also describe two novel genes that are involved in the controlling the spread of DNA methylation in Neurospora.
Centromeres of filamentous fungi
M. Freitag, Lanelle R. Connolly and Kristina M. Smith. Dept. of Biochemistry and Biophysics, Center for Genome Research and Biocomputing, Oregon State University, Corvallis, USA
Centromeres are complex structures composed of DNA, proteins and RNA. We still do not fully understand how centromeres assemble, how they are maintained and how they are inherited. Early studies revealed specific centromeric DNA motifs and associated DNA-binding proteins, but this simple model does not hold true outside of budding yeasts. Centromeres are now functionally defined by the presence of a centromere-specific histone H3 variant, CenH3. In addition, a combination of histone modifications differentiates “centrochromatin” from “heterochromatin” and "euchromatin", but a cause and effect relationship between histone modifications and centromere identity has not been established. One key question regards the relative importance of DNA composition vs. epigenetic modifications in marking the regional centromeres of higher eukaryotes. Similarly, the molecular mechanisms that govern centromere assembly and maintenance remain largely obscure, even though numerous proteins have been implicated in these processes. We are using Neurospora crassa and Fusarium graminearum to address these questions. Centromeric regions in Neurospora are of similar complexity as those found in most animals, bridging the gap between simple (e.g., yeasts) and complex eukaryotes, while lacking the ~180-bp tandem repeats typically associated with plant and mammalian centromeres. The F. graminearum genome appears virtually repeat-free, which makes it ideally suited to investigate the relative contributions of DNA composition and epigenetic modifications to centromere identity. We will show evidence for the localization of two centromeric proteins (CenH3 and Cenp-C) in Neurospora and Fusarium, detected by cytological (GFP fusions) and molecular means (ChIP-Sequencing).
Region specific regulation of recombination - How the discovery of MSUD unmasked a troublesome phantom
F.J. Bowring, P.J. Yeadon and D.E.A. Catcheside. School of Biological Sciences, Flinders University, PO Box 2100, Adelaide, South Australia, Australia
rec-1+, rec-2+ and rec-3+ are dominant trans-acting genes that suppress meiotic recombination in specific regions of the Neurospora crassa genome. As an example, up to 1% of progeny from a rec-2 by rec-2 cross experience recombination in his-3 but this falls to about 0.005% when one or both parents carries rec-2+. While our attempt to clone rec-2+ has encountered some difficulty, without Patrick Shiu and Bob Metzenberg’s discovery of meiotic silencing by unpaired DNA (MSUD), it would have been impossible.
RFLP mapping after a long walk along chromosome V indicated that rec-2+ resides in a 10kb stretch of DNA. A comforting corollary was that this region is absent from rec-2 strains which have in its place a 3kb stretch of unique DNA. Decidedly non-comforting however, were the observations that putting rec-2+ DNA into rec-2 strains failed to yield a rec-2+ phenotype and that deletion of rec-2+ DNA failed to yield a rec-2 phenotype. On top of this, bioinformatic analysis of the 10kb region failed to identify any plausible rec-2+ candidates. Having reached this dead-end, the discovery of MSUD suggested another possibility: rec-2+ does not exist! We wondered if rec-2+ is a phantom that simply appears as a dominant suppressor of recombination because it deprives rec-2 of an opportunity to pair in rec-2+ by rec-2 heterozygotes?
This appears to be so. Disabling MSUD in rec-2+ by rec-2 heterozygotes substantially increases recombination at his-3 and a rec-2 deletion behaves, like rec-2+, as a dominant suppressor of recombination. Indeed, MSUD is responsible for the dominance of all three rec genes. In the absence of MSUD, recombination frequencies at his-1 in rec-1+/rec-1 heterozygotes and at am in rec-3+/rec-3 heterozygotes are indistinguishable from those of rec-1 and rec-3 homozygotes respectively. Thus, rather than rec+ genes producing suppressors of recombination we have shown that the products of rec-1, rec-2 and rec-3 act to promote recombination in specific regions of the genome, a discovery that would not have been possible without the prior discovery of MSUD.
Meiotic silencing in Neurospora
Rodolfo Aramayo. Texas A&M University
In Neurospora, if a segment of DNA is not present on the opposite homologous chromosome in meiosis, the resulting "unpaired" DNA segment is targeted for silencing. This situation occurs when a DNA element gets inserted at a particular chromosomal position (e.g., a situation akin to the "invasion" of a genome by transposable DNA elements). It can also occur when a normal region gets deleted. In both situations, the resulting loop of "unpaired" DNA activates a genome-wide "alert" system that results in the silencing not only of the genes present in the "unpaired" DNA segment, but also of those same genes if present elsewhere in the genome, even if they are in the paired condition. This phenomenon is called, meiotic silencing and was originally described in Neurospora crassa, but has since been observed in nematodes and mammals. In all these organisms, "unpaired or unsynapsed" regions (or chromosomes) are targeted for gene silencing. We think that meiotic silencing is a two-step process. First meiotic trans-sensing compares the chromosomes from each parent and identifies significant differences as unpaired DNA. Second, if unpaired DNA is identified, a process called meiotic silencing silences expression of genes within the unpaired region and regions sharing sequence identity. We are using a combination of genetics, molecular biology and biochemistry aimed at identifying all the molecular players of the process and at understanding how they work together. In this work we describe the genetic, molecular, cytogenetic and biochemical characterization of key components of the system: Sms-2, Sms-3, Sms-4, Sms-5, Sms-6, Sms-7, Sms-8, Sms-9 and Sms-10. In addition, we describe and discuss how mutants in key genes required for recombination and chromosome pairing are not required for gene-specific meiotic silencing.
Genetic interaction between mutagen sensitive mutants
Hirokazu Inoue. Lab of Genetics, Dept of Regulation Biology, Saitama University, Japan
Mutagen-sensitive mutants have been classified into several groups, based on sensitivity to various mutagens, sensitivity of double mutants, spontaneous and induced mutability, vegetative growth and fertility in homozygous crosses. Recently information of DNA sequencing was added to this characteristic. Major groups are excision repair, recombination repair, post-replication repair, damage checkpoint and mismatch repair. Some mutants did not belong to any groups or belong to plural groups in epistasis interaction. Also some combinations resulted in synthetic lethal. We first described synthetic lethal relationship between uvs-3 and uvs-6 20 years ago. What does “synthetic lethal” mean? In this talk I introduce characters of the mus-16 mutant that is highly sensitive to MMS, but not to IR and UV, and is synthetic lethal in combination with some DNA repair-deficient mutants. As mus-16 was not epistatic to other DNA repair mutations, the repair group belonging to was not determined. We cloned this mus-16 gene from Linkage Group V genomic library. The inferred MUS-16 protein did not show any prominent motifs, but it was homologue of Rtt109 of Saccharomyces cerevisiae. Function of MUS-16 was discussed.
Metzenberg Award Lecture
Changing Times.
Jay C. Dunlap. Department of Genetics, Dartmouth Medical School, Hanover, NH 03755
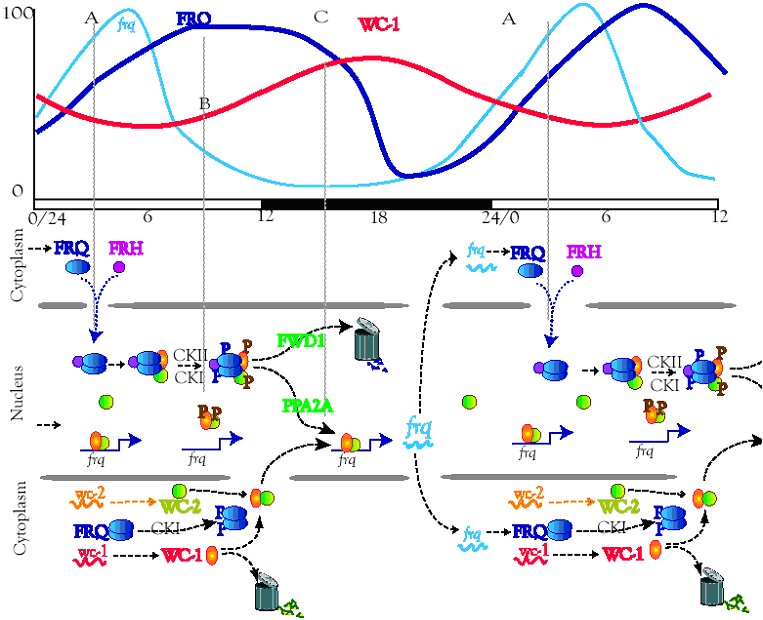
The formal part: Transcription/ translation feedback loops involving
frq are central to circadian rhythms in Neurospora. The transcription
factors WC-1 and WC-2 activate frq expression in a circadian and light-
dependent manner by binding to the Clock Box in the frq promoter. In
cell extracts, WC-1 has always been found complexed with WC-2, so
we expected ChIP experiments to show WC-1 and WC-2 entering and
leaving the Clock Box as a unit at phases associated with increasing and
decreasing frq expression. ChIP, however, indicates that the WC
proteins do
 not act solely as an obligate complex: in vivo binding of
WC-2 to the frq promoter occurs in a rhythmic fashion coincident with
the peak in frq transcription, whereas WC-1 is bound continuously (Belden et al., 2007). Nuclease
accessibility experiments show chromatin rearrangements at frq; a nucleosome occupying the region
near the Clock Box is moved/disassembled in a rhythmic fashion. Deletion of all 19 genes encoding
ATP-dependent chromatin-remodeling enzymes revealed only 2 genes, clockswitch (csw-1 a homolog
of the yeast Fun30, mouse Etl1 and human SMARCAD genes) and chd-2 (a homolog of the
mammalian mi-2 and yeast Chd1/2 genes) required for normal clock function; CSW-1 localizes
rhythmically to frq, suggesting it is required at specific times for the sharp transition from the
transcriptionally active to the repressed state. These data provide a first picture of events happening
at the frq promoter that are important for its essential, correctly timed daily expression.
not act solely as an obligate complex: in vivo binding of
WC-2 to the frq promoter occurs in a rhythmic fashion coincident with
the peak in frq transcription, whereas WC-1 is bound continuously (Belden et al., 2007). Nuclease
accessibility experiments show chromatin rearrangements at frq; a nucleosome occupying the region
near the Clock Box is moved/disassembled in a rhythmic fashion. Deletion of all 19 genes encoding
ATP-dependent chromatin-remodeling enzymes revealed only 2 genes, clockswitch (csw-1 a homolog
of the yeast Fun30, mouse Etl1 and human SMARCAD genes) and chd-2 (a homolog of the
mammalian mi-2 and yeast Chd1/2 genes) required for normal clock function; CSW-1 localizes
rhythmically to frq, suggesting it is required at specific times for the sharp transition from the
transcriptionally active to the repressed state. These data provide a first picture of events happening
at the frq promoter that are important for its essential, correctly timed daily expression.
That’s the formal part, which I expect I’ll get to. Beyond this watch for invocation of Isaac Newton, Bob Metzenberg, David Botstein, the 1st Congregational Church in Thetford, our kitchen, my lab design, and whatever else gets written in and for which there is time.
Session II: Clocks and Light
Circadian Output Pathways in Neurospora.
Deborah Bell-Pedersen. Center for Research on Biological Clocks and Department of Biology, Texas A&M University dpedersen@mail.bio.tamu.edu
Organisms from bacteria to humans use a circadian clock to control daily biochemical, physiological, and behavioral rhythms. We are using the model organism Neurospora crassa to study the circadian clock and its output pathways. Using microarrays we found that ~20% of Neurospora genes are under control of the circadian clock system at the level of transcript accumulation, and that the bulk of the clock-controlled mRNAs have peak accumulation in the late night to early morning. These data suggest the existence of global mechanisms of rhythmic control of gene expression. Consistent with this idea, we found that the Neurospora OS pathway, a phosphorelay signal transduction pathway that responds to changes in osmotic stress, functions as an output pathway from the Neurospora clock system that regulates daily rhythms in expression of downstream genes. Hijacking conserved signaling pathways by the circadian clock provides a new paradigm for global rhythmic control of target genes of the pathway.
Functional Characterization of the White Collar Complex
Tobias Schafmeier, Astrid Schäfer and Michael Brunner. University of Heidelberg Biochemistry Center, INF328, 69120 Heidelberg, Germany
The Neurospora circadian clock protein Frequency (FRQ) participates in interconnected negative and positive feedback loops.
In the negative loop FRQ it inhibits its own transcriptional activator, the White Collar Complex (WCC), and in the positive loop it supports expression of WCC. These apparently contradictory functions are coordinated in a spatial and temporal fashion. Newly synthesized FRQ recruits CK1 and supports phosphorylation of WCC in the nucleus. This leads to inactivation of the transcription factor. Maturation of FRQ causes its progressive accumulation in the cytosol. When high amounts of cytosolic FRQ have accumulated WCC becomes hyperphosphorylation and redistributes from the nucleus into the cytosol. Our data suggest that WC proteins rapidly shuttle between cytosol and nucleus. FRQ affects the subcellular localization of the WCC by controlling its phosphorylation status.
Genome-wide analysis of light induced gene expression and the role of CK2 in temperature compensation.
Jennifer Loros, Chen-Hui Chen, Arun Mehra, Mi Shi, Jay C. Dunlap. Departments of Biochemistry and of Genetics, Dartmouth Medical School, Hanover, NH 03755 USA
Biological rhythms provide organisms with the ability to anticipate environmental changes arising from the Earth's rotation. The genetically determined ability to gauge time of day in concert with the perception of environmental light represents one of the most widespread and closely coupled forms of cellular, tissue and organismal regulation across a broad range of taxonomic groups. Neurospora has played a pioneering role in the description of the molecular basis of both photobiology and circadian rhythmicity. Neurospora has numerous chromophore-binding photoreceptive molecules capable of detecting changes in light fluence and wavelength via the harvesting of photons and thereby allowing immediate signaling of important environmental changes. Genome-wide analysis of changes in transcript abundance has revealed a hierarchical cascade of light regulated responses. Under vegetative conditions most light-inducable genes were found to be dependant on the White-collar 1 and 2 proteins. Additionally, correspondence between timing of expression and gene function was also observed. This work has allowed identification of a cis-acting motif and dedicated transcription factor involved in the control of a specific subset of late light responses. A defining characteristic of all clocks is the ability to maintain constant periodicity over a range of environmental changes including temperature and metabolic rate. Mutations in two Neurospora clock mutants, prd-3 and chr, that result in both period and temperature compensation defects identify subunits of the CK2 enzyme. The prd-3 mutation is in CK2-alpha while chr is defective in CK2-beta. An inducible wild-type CK2-beta subunit that allows graded expression results in temperature compensation phenotypes ranging from anti-compensation to a near wild-type compensation that corresponds to the level of expression of the wild-type protein. The clock protein FRQ is identified as a direct substrate of CK2. FRQ stability is shown to increase with increased temperature in CK2-beta hypomorphs but not with another kinase, CK1, involved in clock function. This suggests that different types of FRQ phosphorylation can lead to changes in either period length alone or period length and compensation.
Neurospora mutants altered in their regulation of transcription by light
Laura Navarro-Sampedro, Maria Olmedo, Carmen Ruger-Herreros and Luis M. Corrochano. Departamento de Genetica, Universidad de Sevilla, Spain
Light activation of transcription in Neurospora is transient due to photoadaptation. The gene con-10 of Neurospora is expressed during conidiation and after illumination of vegetative mycelia. Photoactivation of con-10 is transient and disappears after two hours of light. We have used a N. crassa strain bearing a translational fusion of con-10 to a selectable bacterial gene conferring hygromycin resistance (hph). This con-10::hph fusion is preceded by the con-10 regulatory region. Growth of this strain was shown to be sensitive to hygromycin, upon continuous culture in the light. Five mutants were isolated that were resistant to hygromycin when cultured under constant light. Three of these mutant strains displayed elevated, sustained, accumulation of con-10::hph mRNA, during continued light exposure. This behavior suggests that they bear mutations that reduce or eliminate the presumed light-dependent repression mechanism that normally blocks con-10 transcription upon prolonged illumination.
The gene rco-1, encoding a putative gene repressor, and the gene rco-3, encoding a putative glucose sensor, are required for the repression of con-10 in vegetative mycelia. In addition, we have observed that rco-1 and rco-3 mutants have an enhanced and sustained photoactivation of con-10 and con-6, a phenotype they share with vivid mutants. We propose that VVD, RCO-1, and RCO-3 participate directly or indirectly in the mechanism of adaptation to light.
Does circadian clock matter for Neurospora crassa in nature?
Kwangwon Lee. Cornell University, Ithaca, NY.
Neurospora crassa has been a superb model organism for revealing the molecular nature of circadian clock regulation in filamentous fungi. However, there is, as yet, limited knowledge regarding the ecological significance of N. crassa clock regulation. To understand the quantitative nature and the ecological significance of the clock phenotype in nature, we focused on two research areas; natural variation and QTL (Quantitative Trait Loci) analysis. We found that there is significant natural variation in clock phenotypes and in the known molecular components, WC-1, WC-2, FRQ, and VVD. We focused our study on WC-1, a blue-light receptor and a core circadian clock component. By statistical tests and genetic analysis, we have shown that molecular variation at the amino-terminal activation domain, NpolyQ, in WC-1, contributes to the circadian period variation. Since WC-1 plays a key role in light perception and circadian clock period, we suggest that the molecular variation in WC-1 fine tunes the clock function for ecological adaptation. However, the real question is “does clock regulation matter for fungi in nature?” We addressed this question using two different experimental approaches: 1) comparing fitness of two different strains, wild type and a wc-1 knockout strain, under ecologically-relevant conditions, and 2) measuring fitness of strains with extreme circadian clock period stains. Our data suggest that clock impacts the fitness of an organism. To have a comprehensive understanding of genetic determinants of the circadian clock phenotypes, we preformed QTL analyses on three different populations. We identified 30 QTL for clock phenotype that were not linked to previously known clock genes. Characterizing these QTL will reveal novel insights on quantitative variation of circadian regulation.
White Collar Complex (WCC): acetylation and evolution
B.Grimaldi, A.Brenna, P.Filetici and P. Ballario. Dip Genetica and Mol .biol, UNI and IBPM, CNR, Rome. Italy
Blue light–induced transcription is mainly regulated by the White Collar complex (WCC) in Neurospora and in the other filamentous fungi so far investigated. The white collar-1 (wc-1) and wc-2 genes encode for zinc-finger – PAS trascription activators which constitute the functional core of this complex. When activated by a light stimulus, the WCC drives the transient-transcriptional activation of several photo-regulated genes involved in different physiological processes.
Recently, we have shown that the activation of light-regulated genes by WCC is coupled to light-dependent changes in histone acetylation at their promoters (Grimaldi et al., 2006), indicating that transcription-permissive chromatin states are dynamically established in a light-specific manner.
Indeed, upon a light pulse the promoters of photo-induced genes (i.e. al-3 and vivid) appear acetylated in the residue K14 of histone H3. As the light-driven transcriptional activation, H3 K14 acetylation is transient, depends on WC-1 and follows the RNA kinetic. The enzyme responsible for this chromatin remodelling is NGF-1 (Neurospora Gcn- Five-1), the homologous of S.cerevisiae Gcn5p, a member of the GNAT family of lysine acetyl transferases coactivators. Notably, the structure of WC-1 proteins resembles that of the vertebrate Nuclear Receptor factors (NRs) which interact with transcriptional co-activators (i.e. GCN5, PCAF, p300) upon activation by a stimulus (steroid hormone for NRs, light for WCC). Consistent with this analogy, we demonstrated a physical in vitro interaction between WC-1 and yeast Gcn5p. The interaction requires the presence in the carboxy terminal of WC-1 of the consensus sequences corresponding to the already identified region of binding between NRs and co-activators.
In addition, we will present the comparison between Ncwc-1 versus its hortologous found in an“underground living” fungus, showing adaptative mutations of the function of WCC protein domains in other species.
The effects of prd mutations on the FRQ-less oscillator (FLO)
Patricia Lakin-Thomas
Poster 56
Session III: Cell Growth, Morphogenesis and Populations
Neurospora as a model for evolutionary biology
Jason E. Stajich, Thomas J. Sharpton, Christopher Ellison, Christopher Villalta, David Jacobson, N. Louise Glass, Donald O. Natvig, John W. Taylor.
Research using phylogenetics to characterize Neurospora species, including finding new species that now are being described and extending the ranges of some species in the Northern hemisphere, combined with the many available genetic and developmental tools, make the outbreeding species exceptional candidates for studies of natural evolutionary processes. In addition, research published just last year has initiated the era of experimental, evolutionary studies in Neurospora. Most recently, genome sequencing of N. discreta and N. tetrasperma has opened the door to comparative genomics as a tool to study evolution. We have applied the power of comparative genomics to problems of assembly and annotation that remained for one of the first and best-studied fungal genomes, N. crassa. With improved assemblies and annotation for the three Neurospora genomes, and with genomes for close outgroup species Podospora anserina and Chaetomium globosum, we now are able to investigate gene content, gene family expansion and contraction, synteny and rearrangement, and the effects of selection.
Where is calcium sequestered within cells, and how does it get there
Barry Bowman, Marija Draskovic, Emma Jean Bowman. Department of Molecular, Cell and Developmental Biology. University of California, Santa Cruz CA 95060 bowman@biology.ucsc.edu
Calcium is an important signaling molecule in all cells. In fungi calcium has been proposed to play a key role in polar growth, although we know little about the mechanisms by which calcium is sequestered and released. Analysis of the Neurospora crassa genome led to the identification of at least 5 genes that encode putative calcium transporters (nca-1, nca-2, nca-3, pmr, and cax). We have characterized the phenotypes of strains with mutations in these genes. To investigate the cellular locations of the transporters we made use of both GFP and dsRED tags. Marker enzymes for the vacuolar network, Golgi, ER and nuclei have also been tagged with GFP and dsRED. Analysis of the fluorescent-tagged proteins in living cells shows that the organelles are part of a dynamic system of small vesicles and tubules. The data also suggest that the nuclear envelope may be a significant part of the ER in N. crassa. Different calcium transporters appear to be located in the ER, the vacuolar network and the Golgi.
An Analysis of the Microtubule-Binding Domain of Neurospora Dynein
D.S. Razafsky, J. Ma, R. Schnittker, D. Madole, S.J. King and M. Plamann. University of Missouri-Kansas City, School of Biological Sciences, Division of Cell Biology and Biophysics, Kansas City, MO 64110
Cytoplasmic dynein is a large, microtubule-associated motor complex that facilitates minus-end-directed transport of various cargoes. Using the model organism Neurospora crassa, we utilized a genetic screen to isolate mutants defective in dynein functions. Here we report on two mutations, one within the microtubule-binding domain (MTBD) of the dynein heavy chain and another in the first coiled-coil of the MTBD. In a suppressor screen we found that dynein MTBD mutations are suppressed only by intragenic mutations. Interestingly, the two MTBD mutations discussed here suppress each other’s defects in hyphal growth and nuclear distribution. We purified recombinant MTBD fragments from these strains and determined that the single amino acid mutations alter microtubule-binding frequency in opposite manners. When both mutations were present the frequency of microtubule binding was restored to near wild-type levels. To study the underlying basis of these mutations we tagged the dynein intermediate chain and purified the native motor. Using an in vitro bead motility assay we determined that N. crassa dynein moves with an average velocity of ~2.0 um/s. In the presence of either single amino acid mutation we observed a decrease in the average velocity and distance of dynein translocation. When we examined strains that contain both mutations the velocity was partially restored. Together these data indicate that opposing defects in different domains of dynein heavy chain can compensate for one another.
Regulation of polar tip extension by Rho-type GTPases and COT1 kinase signaling
Nico Vogt and Stephan Seiler. Institute for Microbiology and Genetics; University Goettingen; Grisebachstr. 8; D-37077 Göttingen; Germany
Regulation of Rho GTPase signaling is critical for cell shape determination and polarity. LRG1, a novel member of the GTPase activating proteins (GAPs) was found to be essential for apical tip extension and to restrict excessive branch formation in subapical regions of the hypha and is involved in determining the size of the hyphal compartments. LRG1 localizes to hyphal tips and sites of septation via its three LIM domains. The accumulation of LRG1 as an apical cap is dependent on active growth and influenced by opposing microtubule-dependent motor proteins dynein and kinesin-1. Genetic evidence and in vitro GTPase assays identify LRG1 as a RHO1 specific GAP affecting several output pathways of RHO1, based on hyposensitivity to the glucan inhibitor caspofungin, synthetic lethality with a hyperactive b1,3-glucan synthase mutant, altered PKC/MAK1 pathway activities, hypersensitivity to Latrunculin A and the suppression of lrg-1 by the over expression of the dominant-negative acting N-terminus of the formin BNI1. The morphological defects of lrg-1 are highly reminiscent to the Ndr pathway mutants cot-1 and pod-6, and genetic evidence suggests that LRG1 functions in parallel with COT1 in coordinating apical tip growth.
Mitochondrial Biogenesis
Frank E. Nargang, E. Laura Sherman, Nancy E. Go, and Jeremy G. Wideman. Department of Biological Sciences, University of Alberta, Edmonton, Alberta.
Mitochondria are composed of roughly 1000 different proteins. The vast majority of these are encoded by nuclear genes, synthesized as mitochondrial precursor proteins on cytoplasmic ribosomes, and imported into the organelle. Virtually all mitochondrial precursor proteins carry targeting information that is recognized by the translocase of the outer mitochondrial membrane (TOM complex), which initiates import of the precursors into mitochondria. The core TOM complex contains five different proteins named after their apparent molecular weights: Tom40, Tom22, Tom7, Tom6, and Tom5. Tom40 is the pore forming component of the complex. We have investigated the role of the three small molecular weight Tom proteins by genetic and biochemical analysis. All three proteins appear to play a role in the stability of the complex, but to different extents. Mitochondria lacking Tom6 and Tom7 are affected in their ability to import mitochondrial precursor proteins. Unexpectedly, the absence of any of the small Tom proteins results in an increased rate of assembly of Tom40 into the TOM complex.
Conidial sex tubes and phytochrome signalling
Nick D. Read & H.-C. Kuo. Institute of Cell Biology, University of Edinburgh, Edinburgh EH9 3JH, Scotland.
Nick@Neurospora.org
A new cell type, the conidial sex tube (CST), that can act as a male fertilizing agent, has been discovered in Neurospora crassa. It is morphologically and physiologically distinct from germ tubes and conidial anastomosis tubes (CATs) that also arise from conidia, and under separate genetic control. CSTs are characterized by being long, thin, straight and unbranched, and they do not avoid or grow towards each other. The surface texture and chemistry of CSTs is different to that of germ tubes and CATs. CSTs require the presence of the peptide sex pheromone of opposite mating type to be induced. Nuclear division in CSTs is arrested. Besides being stimulated by sex pheromone, CST induction is also regulated by red light and this is phytochrome-mediated. Neurospora crassa possesses two phytochromes: PHY-1 and PHY-2, and they play contrasting and mating type dependent roles in the photoregulation of CST induction
Genetic and functional characterization of three K+ transport systems in Neurospora
Alberto Rivetta
Poster 3
Identification and partial characterization of Neurospora anastomosis mutants
Stephen Free
Poster 7
Session IV: Gene Regulation and Cell Signaling
Investigation of functional domains of RRG-1, a response regulator that regulates the OS-2 MAPK cascade
Carol Jones
Poster 48
Post-transcriptional control of gene expression in Neurospora
Matt Sachs. Texas A&M University
Fungal mRNAs containing upstream open reading frames (uORFs) can be subject to post-transcriptional regulation. First, translation of the uORF can modulate translation of the main reading frame specified in the mRNA. Second, the uORF termination codon can be recognized as a “premature” termination codon by the machinery responsible for nonsense-mediated mRNA decay (NMD). Fungal mRNAs specifying the small subunit of arginine-specific carbamoyl phosphate synthetase contain a uORF specifying the evolutionarily conserved arginine attenuator peptide (AAP). The synthesis and amino acid sequence of the AAP are critical for cis-acting Arg-specific negative regulation of translation. In cell-free translation systems, AAP-mediated regulation depends on the AAP's ability to stall ribosomes at the uORF termination codon in response to Arg. Certain single missense mutations eliminate ribosome stalling in vitro and Arg-specific regulation in vivo and in vitro. The S. cerevisiae CPA1 and N. crassa AAPs are specified by uORFs whose start codons are in relatively poor translation initiation contexts, resulting in much leaky-scanning past the uORF. Furthermore, when Arg levels are low, there is no stalling of ribosomes on the uORF even when it is translated. However, when Arg is high, ribosomes that have synthesized the AAP stall, and this stalling blocks other scanning ribosomes from initiating translation at the downstream start codon. In addition to translational regulation, CPA1 expression is naturally regulated at the level of mRNA stability via a mechanism involving nonsense-mediated mRNA decay (NMD). Arg-regulated stalling at the CPA1 uORF stop codon triggers NMD and the data suggest a simple model that controlling the density of ribosomes at the uORF termination codon modulates NMD. Analyses of N. crassa arg-2 in isogenic NMD+ and nmd- strains indicate that its levels are also controlled by NMD. We are developing methods to analyze N. crassa mRNA half-life to determine directly how the stability of arg-2 and other mRNAs is controlled, and to determine which trans-acting factors contribute to this control. These studies provide a basis for a general understanding of how ribosome occupancy of a uORF termination codon controls both translation and mRNA stability in eukaryotes.
Regulation of the RNAi pathway in Neurospora
Heng-Chi Lee
Poster 43
The ylo-1 gene encodes an aldehyde dehydrogenase responsible for the last oxidative reaction in the Neurospora carotenoid pathway
Alejandro Fernández Estrada
Poster 1
Trans-species activity of the Neurospora crassa ribonucleotide reductase incompatibility domain
Robert P. Smith, Fuad Tanha, Kenji Wellman, Leila Hadari and Myron L. Smith. Department of Biology, Carleton University, Ottawa, Ontario, Canada
In Neurospora crassa, nonself recognition occurs during vegetative growth. Cell fusion may result in the formation of heterokaryons, cells that contain more than one genetically distinct nucleus. Viability of heterokaryotic cells is governed by at least 11 het (heterokaryon) genes. The het-6 locus contains two genes one of which, un-24, has a dual function as it also encodes the large subunit of a class I ribonucleotide reductase (RNR). un-24 has two alleles, Oakridge (OR) and Panama (PA), which differ in their carboxy termini. Cells that contain both OR and PA, either as heterokaryons or partial diploids, are non-viable or grow slowly with an aberrant morphology. Extended periods of incompatible colony growth result in a phenomenon called 'escape,' a sudden shift to wild-type growth rate and morphology. Recently, this incompatibility reaction was demonstrated to occur when fusion constructs expressing 63 amino acids of the un-24PA C-terminus (hygunPA) were transformed into un-24OR strains.
To further our understanding of this system, we transferred un-24 constructs into Saccharomyces cerevisiae. Several lines of evidence suggest that hygunPA is toxic to yeast. Expression of hygunPA at low levels results in a characteristic incompatible phenotype that includes sensitivity to physical and chemical stressors, an extended lag phase and interruption of the cell cycle, and an aberrant, granulated morphology. Western analysis with RNR antibodies shows that when hygunPA is expressed at low-levels there is a decrease in oxidized RNR and an increase in reduced RNR. During this presentation, we present evidence that the un-24 incompatibility system functions in yeast in a similar fashion to N. crassa. In addition, we will demonstrate how S. cerevisiae can be used to further characterize fungal incompatibility systems.
Plasmid-induced senescence in Neurospora crassa triggers mitochondrial-nuclear communication
Jack Kennell
Poster 11
Characterization of the Neurospora VS ribozyme in vivo
Andrew Keeping
Poster 27
New features over the expression of Neurospora crassa genes in response to extracellular phosphate levels and pH
Fabio Squina
Poster 38
Session V: Genomics and Methodology
Serine-Threonine Kinases and Neurospora Biology
Gloria Turner and Katherine Borkovich. University of California, Los Angeles and University of California, Riverside
Serine-threonine protein kinases are well-known for their involvement in critical signal transduction pathways regulating growth, development and environmental responses in eukaryotes. In order to address the roles of these proteins in Neurospora biology, we attempted to create knockout mutants in all serine-threonine kinase-encoding genes. A total of 89 serine-threonine protein kinases were annotated in the Neurospora crassa genome sequence. Heterokaryotic transformants were obtained for all 89 genes, but we were only able to isolate viable ascospore progeny for 60 kinases. Phenotypic analysis of the viable mutants revealed ~40% exhibited defects in the sexual cycle. Of these, 15 mutants did not produce protoperithecia, while another 10 mutants had defects in perithecial development or ascospore shooting/production. Approximately 19% of the mutants had asexual developmental defects, with the most severe phenotype, the complete absence of conidia, found only in the female-sterile kinase mutants. A total of 27% of the mutants exhibited abnormal growth rates, measured as basal hyphal extension over 72 hours. Of interest, all but one of the female-sterile mutants are included in this mutant phenotype category. In conclusion, serine-threonine kinases were shown to play an important role in growth and asexual and sexual development in N. crassa, and a small group of genes were essential for all three processes.
Neurospora Functional Genomics Projects: Annotation
Heather M. Hood1 and Matthew S. Sachs2. 1Oregon Health & Science University, Beaverton OR 97006; 2Texas A&M University, College Station, TX 77843
Refinement of the Neurospora crassa genome annotation is proceeding on multiple fronts. Published scientific literature from 2001 through the present was reviewed; data were extracted and added to the N. crassa database. To date, nearly 1000 articles were reviewed and over 500 were associated with specific genes in the Broad Institute database
(http://www.broad.mit.edu/annotation/genome/neurospora/Home.html). Information about cloned genes contained within the Neurospora Compendium (Perkins, Radford, and Sachs, 2000) was curated; data from the large-scale genome annotation paper (Borkovich et al., 2004) were also added to corresponding loci. All known and predicted peptides were functionally analyzed using the Gene Ontology (GO) Consortium’s BLAST server. Functional information acquired from these GO BLAST results were subjected to stringent criteria before inclusion in the N. crassa database. Over 1000 genes have curated annotations ascertained from these literature and functional analyses. Finally, each of the 251 contigs were individually inspected a minimum of three times, resulting in over 25% of the predicted genes being manually annotated. When possible, intron/exon boundaries were confirmed or corrected, and untranslated regions were extended using existing and newly sequenced cDNA libraries that are a part of this functional genomics project. The incorporation of data from multiple literature sources, functional analyses, and manual curation of gene models has greatly expanded and refined the information available to the research community from the N. crassa database.
Dissecting colony development of Neurospora crassa using mRNA profiling and comparative genomics approaches
Takao Kasuga and N. Louise Glass. Department of Plant and Microbial Biology, University of California, Berkeley 94720-3102
Colony development, including hyphal extension, branching, anastomosis, and asexual sporulation, is a fundamental part of the lifecycle of filamentous fungi, yet genetic mechanisms underlying these phenomena are poorly understood. We conducted transcriptional profiling during colony development of a filamentous model fungus, Neurospora crassa using 70-mer oligonucleotide microarrays. First, six sections of defined age groups were excised from a 27 hour old colony and subsequently Bayesian inference of relative mRNA expression levels was made. A functional category database was then used to identify biological processes participating during the developmental stages. Genes involved in polar growth and cellular signaling were enriched for expression at the periphery of the colony. The middle part of the colony was engaged in protein synthesis and energy production. In the older part of the colony, genes for proteins and peptide degradation, and unclassified proteins were active. A cross-examination of the N. crassa dataset with a published dataset of Aspergillus niger revealed shared patterns in the spatiotemporal regulation of gene orthologs during colony development. At present less than 50% of N. crassa genes have functional annotations, which impose the chief limitation on data analysis. We developed an alternative method by incorporating an evolutionary approach. First, a phylogenetic distribution of each of the N. crassa genes was used to classify genes into mutually exclusive lineage specific (LS) groups: (1) Eukaryotes/Prokaryotes (Euk/Prok)-core, (2) Dikarya-core, (3) Ascomycota-core, (4) Euascomycetes (Euasco)-specific, and (5) N. crassa-orphans. We then cross-examined mRNA profiles with the LS groups. An enrichment of Euasco-specific genes was detected in the colony periphery. On the other hand, Euk/Prok-core and Dikarya-core genes were enriched in the middle part of the colony. In the order part of the colony, where the region was in the early stage of conidiation, an enrichment of N. crassa-orphans was observed.
Construction and Characterization of New cDNA Libraries from Neuorospora Mauriceville.
Meray Basturkmen1, Junhuan Xu2, Mary Anne Nelson2, Chinnappa D. Kodira3, Bruce Birren3, Matthew S. Sachs1. 1Texas A&M University, Department of Biology, College Station, TX, 2University of New Mexico, Department of Biology, Albuquerque, NM, 3The Broad Institute, Cambridge, MA
Neurospora crassa cDNA libraries were constructed from the Mauriceville Mat A strain (FGSC# 2225). Libraries were prepared from cells grown under 13 different conditions including growth in Vogel’s and Westergaard’s media, heat shock, oxidative stress, osmotic stress, glucose deprivation, nitrogen deprivation and sexual induction. Cells were extracted for total RNA and poly(A) RNA was purified by oligo-dT cellulose chromatography. First strand cDNA was synthesized at high temperature to minimize problems arising from RNA secondary structure. The Gubler and Hoffman RNAse H method was used for second strand synthesis. Double-strand cDNA was directionally cloned into the UNIZAP XR vector using EcoRI and XhoI sites. Lambda libraries were amplified and excised to obtain single-strand phage containing pBlueScript for sequencing. Each individual library of amplified lambda phage was also deposited at the Fungal Genetics Stock Center. Test sequencing provided evidence for more than 700 new genes with a high rate of full-length cDNA sequences. Deeper sequencing and the analysis of the libraries are continuing.
Cross-kingdom patterns of alternative splicing and splice recognition
Abigail Manson McGuire*, Matthew D. Pearson*, Daniel E. Neafsey, and James E. Galagan (*these authors contributed equally)
We have performed a comprehensive survey of alternative splicing across 42 eukaryotes – including 14 fungi - to gain insight into the recognition of spliceosomal introns. The observed variations in transcript splicing reveal potential differences in how eukaryotes recognize intronic splice sites. Intron recognition by intron definition (ID) implies a greater number of shorter retained introns (RIs) whereas intron recognition by exon definition (ED) implies a greater number of shorter cassette exons (CEs).
All eukaryotes we examined exhibit RIs, which appear more frequently than previously thought. CEs are also present in all kingdoms and most of the organisms that we studied. We observe that the ratio of CEs to RIs varies substantially among kingdoms, while the ratio of competing 3’ acceptor and competing 5’ donor sites remains nearly constant. In addition, we find the ratio of CEs to RIs in each organism correlates with the length of its introns.
In all 14 of the fungi that we examined, as well as in most of the 9 protists, RIs far outnumber CEs. This trend was observed for both ascomycetes and basidiomycetes and was independent of the number of introns present in the organism. This differs from the trend seen in the 13 multicellular animals in our analysis, where CEs occur more frequently. The 6 plants we analyzed have substantial amounts of both CEs and RIs. Our results suggest that most extant eukaryotes are capable of recognizing splice sites via both ID and ED, although ED predominates in multicellular animals and ID is more common in fungi and most protists.
Neurospora Lights Up!
Van Gooch†, Arun Mehra§, Luis Larrondo§, Julie Fox†, Jennifer Loros§ and Jay Dunlap§. †Division of Science and Mathematics, University of Minnesota—Morris, Morris, MN 56267, and § Department of Genetics, Dartmouth Medical School, Hanover, New Hampshire 037552.
We have synthesized a firefly luciferase gene to be compatible with the codon usage of Neurospora crassa. The third codon position of the native firefly (Photinus pyralis) luciferase gene is rich in adenine and thymine while usage tables show that Neurospora tends to have a preference for cytosine and guanine in that position. The optimized gene was designed to recognize these Neurospora preferences while the eventual amino acid sequence of the luciferase would be identical to the native firefly luciferase. A total of 426 individual nucleotide changes were made out of the 1653 base pair sequence of the native firefly luciferase. Eighty-six oligonucleotides of 40 bp in length with 20 mer overlaps were purchased and assembled into a gene using assembly PCR. The gene was designed so that promoters of choice could be easily inserted. Initially we created strains using a segment of the frq promoter (known to be integral in the circadian mechanism) and the eas/ccg-2 promoter (known for its vigorous ability to induce translation as well as being controlled by the circadian clock). Neurospora transformed with eas/ccg-2 optimized luciferase construct in the presence of luciferin yields a glow strong enough to be seen by the dark-adapted eye. The bioluminescence from the frq optimized luciferase construct demonstrates all of the classic properties of Neurospora circadian rhythms. This new system allows for detailed and high volume monitoring of the molecular level circadian oscillator in Neurospora. Detailed phase response curves for light and temperature have been generated. We believe the codon optimized luciferase gene should be a valuable tool to all researchers of Neurospora and fungal genetics and that the concept of codon optimization is an important consideration for those looking for high protein expression.
Expression of the red fluorescent protein mCherry During the circadian rhythm of Neurospora crassa
Ernestina Castro-Longoria
Poster 8
Development of Neurospora host strains with reduced codon bias for elevated protein expression
Christina Chung
Poster 60